1. What is the definition of SOP?
What are contents required for SOP? Information should master document carry on every page not just one of the pages to meet GMP?
SOPs required for equipment?
List SOPs required in QA department
· SOPs are detailed written instructions for the
operations routinely performed in the course of any activities associated with
pharmaceutical manufacturing.
·
A written authorized procedure which gives
instructions for performing operations not necessarily specific to a given
product / material, but of a more general nature the equipment preventive
maintenance and cleaning; recall of products; purchasing; cleaning of premises
and environmental control; sampling and inspection etc.
·
These are guidelines which describe how the
activity is to be performed. To achieve uniformity of results by each
individual, it is mandatory to follow these guidelines.
·
SOP is like a “TELL and SHOW” concept. Tell –
means to establish and teach how the activity is to be carried out. Show –
means to provide the documented proof for the activity carried out.
Contents
of SOP
- Objective/Purpose,
- Scope
- Responsibility
- Accountability
- List of formats/Annexure
- Procedure
- Abbreviations
- Reference
- Revision History
Information should
master document carry on every page not just one of the pages to meet GMP
- Page number
- Document reference number
- Authorizing signatures
SOPs
required for equipment
- Operation
- Cleaning
- Preventive maintenance/ Calibration
- Sampling procedure
List SOPs
required in QA department
·
SOP for SOP
·
SOP for format preparation,
·
Change control
·
Deviation
·
Non-conformance products,
·
Market complaints
·
Product recall
·
Returned goods
·
Vendor qualification
·
Preparation of BPCR & MPCR
·
Assigning of Mfg. date & Expiry date
·
Annual product review
·
Corrective action & preventive action
·
Process validation, cleaning validation
·
Equipment qualification
·
Glossary of terms, document control
·
Review of BPCR & analytical test report
·
Batch numbering system
·
Labeling practice
·
Personnel training
·
BPCR issue and retrieval
·
Batch release
·
Self-inspection (internal audit)
·
File numbering system
·
Preparation of organo-gram
·
Preparation of COA
·
Specimen signatures
·
Reprocess & rework of intermediates / API
·
Job responsibilities
·
Technology transfer
·
Measurable quality objectives etc.
[ads id="ads1"]
2. What is the
Batch production and control record (BPCR) & Master Production &
control record (MPCR)?
Batch
production and control record (BPCR)
BPCR are
prepared for each intermediate and API and include the complete information relating
to the completion of each significant step in the Batch production.
Master
production & control record (MPCR)
To ensure
the uniformity from batch to batch, master production instructions for each intermediate
and API are prepared, dated and signed by one person, immediately checked, dated
and signed by a person in the quality unit.
Content of
the MPCR
- The name of the intermediate or API being
manufactured and an identifying document reference code, if applicable
- A complete list of raw materials and
intermediates designated by names or codes sufficiently specific to identify
any special quality characteristics
- An accurate statement of the quantity or ratio
of each raw material or intermediate to be used, including the unit of measure.
- Where the quantity is not fixed, the calculation for each batch size or rate of
production should be included. Variations to quantities should be included
where they are justified
The production location and major production
equipment to be used
Detailed production instructions, including the:
o
Sequences to be followed
o
Ranges of process parameters to be used
o
sampling instructions and in-process controls
with their acceptance criteria, where appropriate
o
Time limits for completion of individual
processing steps and/or the total process, where appropriate
o
Expected yield ranges at appropriate phases of
processing or time
o
Where appropriate, special notations and
precautions to be followed, or cross references to these
·
The instructions for storage of the intermediate
or API to ensure its suitability for use, including the labeling and packaging
materials and special storage conditions with time limits, where appropriate.
[ads id="ads2"]
3.
What is the
difference between intermediate and drug substance (API)? What is the difference
between drug substance and drug product?
·
Intermediate: A material produced during steps
of the processing of an API that undergoes further molecular change or
purifications before it become an API (Reference: ICH Q7A).
·
API: Any substance or mixture of substances
intended to be used in the manufacturing of a drug (medicinal) product and that
when used in the production of a drug, becomes an API of the drug product.
·
Such substances are intended to furnish
pharmacological activity or other direct effect in the diagnosis, cure,
mitigation, treatment or prevention of disease or to affect the structure &
function of the body (Reference: ICH Q7A).
·
Drug substance (API): Any substance or mixture
of substances intended to be used in the manufacture of a drug (medicinal)
product and that, when used in the production of a drug becomes an active
ingredient of the drug product. Such substances are intended to furnish pharmacological
activity or other direct effect in the diagnosis, cure, mitigation, treatment,
or prevention of disease or to affect the structure and function of the body
(Reference: ICH Q7A).
·
Drug product: The dosage form in the final
immediate packaging intended for marketing (Reference: ICH Q7A).
Quick Searches
4.
What is the difference between GMP & cGMP?
·
GMP: GMP is the
part of Quality assurance which ensures that products are consistently produced
and controlled to the quality standards appropriate to their intended use and
as required by the marketing authorization.
·
GMP are aimed
primarily at diminishing the risks inherent in any pharmaceutical production.
Such risks are essentially of two types:
1. Cross-contamination (in particular of unexpected contamination)
2. Mix-ups (confusion)
·
cGMP: Current
Good Manufacturing Practices. This means any procedure / system adopted by the
manufacturer which proves to be necessary and important for identity, strength
and purity of a product.
[ads id="ads2"]
5.
What is the clean room? What are the
classifications of clean rooms?
·
Clean rooms are
defined as especially constructed, environmentally controlled enclosed spaces
with respect to airborne particulates, temperature, humidity, air pressure, air
low patterns, air motion, vibration, noise, viable (living organisms) and
lighting.
·
Particulate
control includes:
o
Particulate
& microbial contamination
o
Particulate
concentration & dispersion
Ø
Generally clean
rooms are classified in to the following types as per different guidelines:
- ·
Schedule M:
Grade A, Grade B, Grade C, Grade D
- ·
USFDA (US
209E): Class 1, Class 10, Class 100, Class 1000, Class 10000, Class 100,000
- ·
WHO 2002: Grade
A, Grade B, Grade C, Grade D
- ·
EU GMP: Grade
A, Grade B, Grade C, Grade D
- ·
ISO 14644-1:
ISO-3, ISO-4, ISO-5, ISO-6, ISO-7, ISO-8, ISO-9
- ·
Britain (BS
5295): Class C, Class D, Class E or F, Class G or H, Class J, Class K
- ·
Australia (AS
1386): 0.035, 0.35, 3.5, 35, 350, 3500
- · Germany (VDI
2083): 1, 2, 3, 4, 5, 6
[ads id="ads1"]
6.
What is the difference between Qualification
and Validation? What is the definition of Validation & Qualification? What are the types of validation?
·
Qualification
is equipment / instrument oriented but validation is process oriented.
·
Validation is
the documented program that provides a high degree of assurance that a specific
process, method or system will consistently produce a result meeting
predetermined acceptance criteria.
·
Qualification
is the action of proving and documenting that any equipment or ancillary
systems are properly installed, work correctly, actually leads the expected
results. Qualification is part of validation, but the individual qualification
steps alone do not constitute process validation.
Types of validation
- Process
validation
- Analytical
method validation
- Cleaning
validation
- Facility
validation
- Utility validation
& software validation
Process
validation and types of process validation
Process
validation is the documented evidence that the process, operated within
established parameters, can perform effectively and reproducible to produce an
intermediate / API meeting its pre-determined specifications and quality
attributes.
Process
validation is three types:
1. Prospective
process validation
2. Concurrent
process validation
3.
Retrospective process validation
Prospective
process validation:
Prospective Process validation shall be carried out
for all the intermediate stages and Active Pharmaceutical Ingredients prior to
the distribution of a new product. [ICH: GMP, EU: GMP, PIC/S: GMP]
Concurrent process validation:
Any validated process undergoes a change either for
the equipment or addition, deletion of a critical manufacturing process step,
scale up or scale down, the same needs to be validated concurrently.
The validation is carried out only after a change
of an existing validated process to support the change made or involve with the
requirements.
Or
A subset of prospective validation in which API
batches are released for distribution, based on extensive testing, before
completion of process validation. Once data from additional batches produced
under replicated conditions show uniformity, the process may be considered
validated
Or
Concurrent validation can be conducted when data
from replicate production runs are unavailable because only a limited number of
API batches have been produced, API batches are produced infrequently, or API
batches are produced by a validated process that has been modified. [ICH: GMP,
EU: GMP, PIC/S: GMP]
Retrospective
process validation:
Validation of a process for a product already in
distribution based upon accumulated production, testing and control data. [ICH:
GMP, EU: GMP, PIC/S: GMP]
[ads id="ads1"]
7. What do you mean by validation protocol and its contents of
process validation?
A written plan stating, how validation will be conducted and
defining acceptance criteria
e.g.: The protocol for manufacturing process identifies process equipment,
critical process parameters, and / or operating range, product characteristics,
sampling, test data to be collected, number of validations runs and acceptance
test results.
Contents
Protocol Approval
· Table of contents
· Objective
· Scope
· Responsibility
· Accountability
· Validation team
· Brief manufacturing process (Description, Flow chart, Reaction scheme)
· Selection of batches
· List of equipment’s used in the manufacturing process
· List of raw materials used in the manufacturing process
· Critical operations with justification
· In-process controls with acceptance criteria
· Sampling & testing plan with frequency
· Stability program
· Data to be complied
· Acceptance criteria
· Intermediate & final products quality & yield
· Stability specification
· Document review
· Conclusion
· Revalidation criteria
[ads id="ads2"]
8. What is
the definition of the procedure?
A documented
description of the operation to be performed, the precautions to be taken, and measures
to be applied directly or indirectly related to the manufacture of an intermediate
/ API (Reference: ICH Q7A).
9. What is the master
document?
Master document
is a formally authorized source document relating to specifications, and / or manufacturing
/ analytical methods, which is protected from un-authorized access or amendment.
· Documents required describing the quality system requirements in the organization.
· Documents required describing the process or product characteristics.
· Documents required by various regulatory agencies as part of compliance to GMP requirements.
· Documents required for legal/ regulatory supports of the organization to meet the local regulations.
· Any other documents required by government / regulatory agency.
Popular Searches
10. What is
documentation?
All the written production
procedures, instructions and records, quality control procedures and recorded
test results involved in the manufacturing of a medicinal product
11. What is the Technology Transfer?
·
In the pharmaceutical
industry, “technology transfer” refers to the processes that are needed for
successful progress from drug discovery to product development to clinical
trials to full scale commercialization or it is the process by which a
developer of technology makes its technology available to commercial partner
that will exploit the technology.
·
To assure the drug quality, it
is desire to make sure 5 W’s and 1 H, that is what1, when2, and why3
information should be transferred to where4 and by whom5 and how to transfer,
then share knowledge and information of the technology transfer each other
between stake holders related to drug manufacturing.
[ads id="ads1"]
12. What are the names of different countries
of GMP guidelines for manufacturing of API?
- · WHO GMP - Geneva
- ·
ICH Q7A – Europe, Japan &
US
- ·
EU GMP - Europe
- ·
MCC – South Africa
- ·
APIC GMP – Active
Pharmaceutical Ingredient Committee (A sector group of CEFIC)
- ·
USFDA GMP – United States of
America
- ·
PIC/S GMP- Germany
- ·
Schedule M – Indian
[ads id="ads1"]
13. What is preventive maintenance?
·
It is periodic inspection and
minor repairs of equipment as per schedule given in the SOP. This enables
smooth operation and long life of the equipment. It also avoids major breakdown
of the equipment during manufacturing of the product.
·
There are two types of
maintenance.
o Preventive maintenance: Schedule maintenance before any break down
of machinery which prevents the machine break down.
o Breakdown maintenance: Maintenance was done after stopping machine
breakdown. Weekly, Monthly, Quarterly, Half yearly and Yearly preventive
maintenance
[ads id="ads1"]
14. What do you mean by “Quality Assurance”?
The sum total
of the organized arrangements made with the objects of ensuring that all APIs are
of the quality required for their intended use and the quality systems are
maintained.
[ads id="ads1"]
15. What are the types of different training
programs?
1. Induction training
2. Job oriented training
3. cGMP training
4. On-going training
[ads id="ads1"]
16. What is cGMP?
Current Good
Manufacturing Practices.
This means any
procedure / system adopted by the manufacturer which proves to be necessary and
important for identity, strength and purity of a product.
[ads id="ads1"]
17. What are the requirements for the
equipment used in the manufacturing of process of API?
Material of
construction used for equipment should not
·
React with component
·
Get corroded, cause rusting
·
Impart any impurities, absorb
·
Should be of appropriate
design, adequate size and have smooth surface.
[ads id="ads1"]
18. How are cGMP implemented?
Training, compliance to SOPs, control on operations, following
procedures / systems, monitoring through compliance audits.
[ads id="ads1"]
19. What is solvent? What are the
classifications of residual solvents?
An organic or inorganic liquid used as a vehicle for the
preparation of solutions or suspensions in the manufacturing of an intermediate
/ API.
Residual solvents are classified into three class based on the
possible risk to human health:
·
Class-I
(Solvents to be avoided)
·
Class-II
(Solvents to be limited)
·
Class-III
(Solvents with low toxic potential)
[ads id="ads1"]
20. What is the difference between
Responsibility and Accountability?
Responsibility: Personnel directly associated with the
implementation of the procedure
Accountability: Person directly associated with the implementation
of the system under which the procedure falls.
21. Write the names of the different
countries regulatory body (Like for India, USA, UK, Australia, South Africa,
Brazil, Hungary, Germany, Philippines etc.)
- India –
Schedule M
- United Status
of America – USFDA (United States Food and Drug Administration)
- Australia – TGA
(Therapeutic Goods Administration)
- United Kingdom
– MHRA (Medicines & Health care products Regulatory Agency)
- South Africa –
MCC (Medicine Control Council)
- Brazil – ANVISA
(Brazilian Health Surveillance Agency or National Sanitary Surveillance Agency)
- Hungary - PIC/S
(Pharmaceutical Inspection Convention or Pharmaceutical Inspection Cooperation
Scheme)
- Germany – NIP
(National Institute of Pharmacy)
- Philippines –
BFAD (Beaureu of Food & Drug)
[ads id="ads1"]
22. What is the abbreviation of MSDS and how
many contents are mentioned & what are those?
MSDS means Material Safety Data Sheet and it contains 16 contents.
Those are given below:
1. Product Identification
2. Composition / Information on Ingredients
3. Hazards identification
4. First Aid measures
5. Firefighting measures
6. Accidental release measures
7. Handling & storage
8. Exposure controls / Personal protection
9. Physical & Chemical properties
10. Stability & Reactivity
11. Toxicological information
12. Ecological information
13. Disposal consideration
14. Transport information
15. Regulatory information
16. Other information
[ads id="ads1"]
23. What is the static electricity?
Denoting / pertaining to electricity which is at rest. The
electricity which is present on surface of a non-conductive body, where it is
trapped from escaping, is called static electricity.
[ads id="ads1"]
24. What is the different types of
Qualifications and write its flow?
Qualifications are as follows:
- Design Qualification,
- Installation Qualification,
- Operational Qualification,
and
- Performance Qualification.
URS/DS -----FAT-----SAT-----DQ-----IQ-----OQ-----PQ
[ads id="ads1"]
25. What is audit/inspection and Why quality
audit? Write different types of audits/inspection?
A planned and systematic examination and check of a system,
procedure or operation in order to monitor compliance with and the
effectiveness of established standards and to allow for improvement and
corrective measures where required.
Quality audit because of:
- To assess the effectiveness of the quality management system
- Assessing conformance
- Investigating problems
- Continual improvement of performance
- Assessing for Registration
- Reducing cost of operation
- Legal requirement
Types:
1. Study/test based inspection
2. Facility based inspection
3. Process based inspection
[ads id="ads1"]
26. Why nitrogen gas used in the
manufacturing area at room temperature and why not other gas?
Because of nitrogen is chemically less reactive and does not react
with other elements at ordinary temperature. It is due to strong bonding in its
molecule.
[ads id="ads1"]
27. What are the different types of
cleanings?
There are three types of cleanings:
- Batch to Batch cleaning
- Periodically cleaning
- Product change over cleaning
[ads id="ads1"]
28. What is blending?
Blending is defined as the process of combining materials within
the same specification to produce a homogeneous intermediate or API.
[ads id="ads1"]
29. What is expiry date & re-test date?
Expiry date
·
The date place
on the container / labels of an API designated the time during which the API is
expected to remain within established shelf life specifications if stored under
defined conditions and after which it should not be used.
Re-test date
·
The date when a
material should be re-examined to ensure that it is still suitable for use. The
period of time during which the drug substance is expected to remain within its
specifications and therefore, can be used in the manufacturing of the drug
product, provided that drug substance has been stored under the defined
conditions.
[ads id="ads1"]
30. What is difference between reprocess
& rework?
Reprocess:
·
Introducing an
intermediate or API, including one that does not conform to standards or
specifications, back into the process and repeating a crystallization step or
other appropriate chemical or physical manipulation steps (e.g., distillation,
filtration, chromatography, and milling) that are part of the established
manufacturing process.
·
Continuation of
a process step after an in-process control test has shown that the step is
incomplete, is considered to be part of the normal process, and is not
reprocessing.
Reworking:
·
Subjecting an
intermediate or API that does not conform to standards or specifications to one
or more processing steps that are different from the established manufacturing
process to obtain acceptable quality intermediate or API (e.g., recrystallizing
with a different solvent).
31. What are deviation &
its types?
Deviation is
departure from the approved instructions /established standards.
There are two
types of deviation and given below:
Controlled /
planned deviation:
·
Any deviation from documented
procedure opted deliberately for temporary period to manage unavoidable
situation or improving the performance of the operations, without affecting the
quality & yield of drug substance and safety of the operations shall be
termed as controlled / planned deviation.
Uncontrolled /
unplanned deviation:
·
Any deviation occurred in
unplanned or uncontrolled manner such as system failure or equipment breakdown
or manual error shall be termed as uncontrolled / unplanned deviation.
[ads id="ads1"]
32. What are change control
and its types?
Change control
is a system that control change by
- Identifying ownership of the
change
- Allowing for review and approval of the
change.
- Preventing changes that could adversely affect
product quality or conflict with registration or regulatory requirement.
- Providing an assessment of change and monitors
the impact of change.
Level 1 (Minor)
are those that are unlikely to have any detectable impact on the quality
attributes of the product.
Level 2 (Major)
are those that are likely to have a significant impact on the quality
attributes of the product.
The type of
reasons for change control
- Regulatory requirement
- GMP implementation /
enhancement
- Quality improvement
- Capacity enhancement
- Introduction of new product in
existing facility
- Cost reduction
- Automation
- Aging of facility
- To manage the unavoidable
situation
- Market requirement
[ads id="ads1"]
33. Definitions
Batch number, Batch contamination
Cross-contamination
Quarantine
Critical process parameters
Mother liquor,
OOSTheoretical yield
Expected yield
CAPA
Installation Qualification (IQ)
Operating Qualification (OQ)
Performance Qualification (PQ)
Batch Number:
A unique
combination of numbers, letters, and/or symbols which identifies a batch (or
lot) and from which the production and distribution history can be determined
Batch:
A specific
quantity of material produced in a process or series of processes so that it is
expected to be homogeneous within specified limits. In the case of continuous
production, a batch may correspond to a defined fraction of the production.
Batch size may be defined either by a fixed quantity or the amount produced in
a fixed time interval.
Contamination:
The undesired
introduction of impurities of a chemical or Microbiological nature, or of
foreign matter, in to or onto a raw material, intermediate, or API during
production, sampling, packaging or repackaging, storage or transport.
Cross-contamination:
Contamination
of a material or of a product with another material or product.
Quarantine:
The status of
materials isolated physically or by other effective means pending a decision on
their subsequent approval or rejection.
Critical
process parameters:
A process parameter whose variability has an
impact on a critical quality attribute and therefore should be monitored or
controlled to ensure the process produces the desired quality.
Mother liquor:
The residual
liquid which remains after the crystallization or isolation processes. Mother
liquor may contain un-reacted materials, intermediates, levels of the API
and/or impurities. It may be used for further processing.
OOS:
Out of
Specification (OOS) results are those results, generated during testing that do
not comply with the relevant specification or standards or with the defined
acceptance criteria.
Theoretical
yield:
The quantity
that would be produced at any appropriate phase of production, based upon the
quantity of material to be used, in the absence of any loss or error in actual production.
Expected yield:
The quantity of
material or the percentage of theoretical yield anticipated at any appropriate
phase of production based on previous laboratory, pilot scale, or manufacturing
data.
CAPA is the
Corrective Action & Preventive Action.
Corrective
Action:
Action taken to
eliminate the causes of an existing non-conformity, defect or other undesirable
situation to prevent recurrence
[Actions taken after the occurrence of a defect
or problem to stop the same from recurrence]
Preventive
Action:
Action taken to
eliminate the causes of potential non-conformity, defect or other undesirable situation
to prevent occurrence
[Actions
initiated before the occurrence of a defect or problem to prevent the same
occurrence].
Installation
Qualification (IQ):
Establishing
high degree of confidence that the equipment as installed is consistent with
manufacture’s requirements and specifications.
Operating
Qualification (OQ):
Establishing a
high degree of confidence that the equipment as installed is able to
consistently operate within established limits and tolerances.
Performance
Qualification (PQ):
Establishing a
high degree of confidence, with appropriate testing that the equipment, under
normal operating conditions, will consistently produce a quality product.
[ads id="ads1"]
34. What is the ICH? Write its
aim/purpose and names of the different parties & different regions?
ICH means
“International conference on harmonization”.
Aim/Purpose:
“Ensure good
quality, safety and effective medicines are developed and registered in the
most effective manner, through harmonization of technical requirements”
Different
Parties:
1. European
commission – European Union (EMEA)
2. European
Federation of Pharmaceutical Industries & Association (EFPIA)
3. Minister of
health, Labor & Welfare, Japan (MHLW)
4. Japan
Pharmaceutical Manufacturers Association (JPMA)
5. US Food
& drugs Administration (FDA)
6.
Pharmaceutical Research & Manufactures of America (PhRMA)
Different
regions:
1. European
Union (EMEA)
2. United
states of America (USFDA)
3. Japan (MHLW)
[ads id="ads1"]
35. Difference between
validation & testing?
Both are not
same. Testing is defined as the identification of errors (difference between expected
& actual results) in a system.
Validation is defined as documented evidence
that a system performance as expected. Validation includes testing but it is
more – for instance, checking the documents for completeness & correctness.
36. Why water is used
extensively as a coolant in heat exchange equipment’s?
Because of the
abundance and high heat capacity, water is used as coolant in heat exchange equipment.
[ads id="ads1"]
37. What are the different
characteristics of the fluid are to be considered while deciding its route in a
heat exchanges?
The following
characteristic of the fluid are to be considered while deciding its route in a
heat exchanger: a) Viscosity b) Fouling c) Corrosiveness d) Pressure
38. When steam distillation
recommended?
1. To separate appreciable quantities of higher
boiling materials.
2. To separate relatively small amounts of
volatile impurity from a large amount of material.
3. Where use of direct-fired heaters is
detrimental to the materials.
4. Where the material is to be subjected to
distillation is thermally unstable or will react with other component
associated with it at the boiling temperature.
5. Where the
material cannot be distilled by in-direct heating even under low pressure because
of the high boiling temperature.
[ads id="ads1"]
39. What is the difference
between instrument & equipment?
Instrument:
A device that
take a physical measurement and displays a value or has no control or
analytical functions.
e.g.: Stop
watch, timers & thermometer.
[A device
<chemical, electrical, hydraulic, magnetic, mechanical, optical,
pneumatic> used to test, observe, measure, monitor, alter, generate, record,
calibrate, manage or control physical properties, movements, or other
characteristics].
Equipment:
A device or
collection of components that perform process to produce result.
[The collective
analytical measurement instruments in conjunction with firmware, assembled to
perform a mechanical process]
[ads id="ads1"]
40. What is HVAC?
·
The HVAC is designed to
circulate the air in the area after passing it over cooling & heating coils
to maintain the required environmental conditions & passing it through the
series of filters to maintain desired cleanliness level in the area.
·
The air in-take and out-take
of the system is designed to maintain certain degree of pressure gradient in
the area as per requirements.
Or
·
HVAC system function is to
condition (heating & cooling), replace (makeup, fresh air, oxygen replacement),
and pressurize (contaminant) and clean (filter) the air in the environment to meet
the required operational conditions.
·
To achieve this objective,
electrical, mechanical & electronic components are arranged in several
configurations such that they produce the expected results.
[ads id="ads1"]
41. What is the meaning of Q, S, E, and M in the ICH?
• “Q” stands for Quality
• “S” stands for Safety
• “E” stands for Efficacy and
• “M” stands for Multi dispensary
[ads id="ads1"]
42. How many guidelines are present in Q & what are those, describe in detail?
In Quality (Q), total 10 guidelines are present. Those are as follows:
1. Q1 - Stability
2. Q2 - Analytical Method validation
3. Q3 - Impurities
4. Q4 - Pharmacopoeia
5. Q5 - Biotechnological quality
6. Q6 - Specification
7. Q7 - Good Manufacturing Practice (GMP)
8. Q8 - Pharmaceutical Development
9. Q9 - Quality Risk Management
10. Q10 - Pharmaceutical Quality System
[ads id="ads1"]
43. How many types of raw material and packing material?
• Raw materials are classified into two types. Those are as follows:
1. Key raw material
2. Other raw material
• Packing materials are classified into two types. Those are as follows:
1. Primary Packing material
2. Secondary Packing material
[ads id="ads1"]
44. Define the Key raw material/ starting material & primary packing material?
Key raw material/starting material:
Starting material shall be defined as that which is
• Incorporated as a significant structural fragment of the API / Drug Intermediate and
• Having significant effect on the Quality and Yield of the product.
• Starting material shall be identified in TDP.
Primary Packing material: Packing material, which come in direct contact with the
API/Intermediate is considered as Primary packing material.
[ads id="ads1"]
45. What is cleaning validation?
Cleaning validation is documented evidence that an approved cleaning procedure will provide equipment which is suitable for processing of pharmaceutical products or APIs.
Cleaning validation is the confirmation of reliable cleaning products so that the analytical monitoring may be omitted or reduced to a minimum in the routine phase.
It describes the validation of cleaning procedures for the removal of contaminants associated with the previous products, residues of cleaning agents as well as the control of potential microbial contaminants.
[ads id="ads1"]
46. What are the sampling techniques used in the cleaning validation?
Swab sampling:
• Areas which are reasonably accessible & hardest to clean can be evaluated, leading to level of contamination or residue per gives surface area.
Rinse sampling:
• Large areas or parts of equipment’s which could not be swabbed should be rinse sampled or directly extracted by solvent.
• Tubes, nozzles, pipes or containers with surface those are not reasonably accessible for direct surface sampling have to be rinsed with solvent.
• In addition, inaccessible areas of equipment that cannot be routinely disassembled can be evaluated.
[ads id="ads1"]
47. What parameters considered during performance qualification of HVAC?
The following parameters are to be considered during the performance qualification of HVAC:
1. Calibration test certificates of instruments
2. Training records of validation team
3. Pressure drop across the HEPA & fine filters
4. Air velocity measurement & calculation of Air changes
5. Integrity test of HEPA filter
6. Differential pressure test
7. Temperature & Relative Humidity test
8. Air flow direction test
9. Cleanliness class verification (Non-viable particle count)
10. Sound level test
11. Light level test
12. Air borne viable particle monitoring
13. Recovery Study
[ads id="ads1"]
48. What are the contents in the BPCR?
BPCR contains the following contents, but not limited:
1. Product Name
2. Stage
3. BPCR Document Number
4. MPCR Reference Number
5. Batch Number
6. Date of Manufacturing
7. Date of Expiry/Re-test
8. Batch release details
9. List of equipment’s used
10. List of raw materials & Quantity with UOM
11. General instructions, Control & Safety instructions
12. Detailed step wise written manufacturing procedures
13. Actual results record for critical process parameters
14. Identity of In-process & Laboratory test results
15. Signatures of person performing details along with supervising details
16. Description of Packaging details
17. Yield calculation
18. Representative of labels for intermediates / raw materials
19. Deviation details
20. Batch starting & completion date
[ads id="ads1"]
49. What is OOT and define?
“OOT” stands for Out Of Trend.
It means any test results obtained for a particular batch that is markedly different the results of the batches in a series obtained using a same validated method.
50. How will you prevent cross-contamination between two different products manufactured in the one production block?
By maintaining the proper pressure differential between the rooms with two Air handling units (if re-circulation) / one Air handling unit (if 100% fresh air)
51. What is limit of Temperature and relative humidity in the pharma area?
Temperature: 25±2˚C & Relative Humidity: 50±5%
52. What is the difference between dedicated and non-dedicated equipment’s?
Dedicated equipment
•
It is used solely for the production of a single product or product line. Concerns over cross-contamination with other products are markedly reduced.
•
Dedicated equipment’s must be clearly identified with the restrictions of use in order to prevent potential errors during cleaning and preparation.
Non-dedicated equipment
•
Where same piece of equipment utilized for a range of products formulations. Prevent of cross-contamination between products becomes the main objective in the cleaning validation effort.
•
Clearly, cleaning non-dedicated equipment’s represents a more significant obstacle to overcome.
[ads id="ads1"]
53. Why three batches consider for the validation?
Because of First one is for information, Second one is for confirmation and Third one is for evidence.
54. If one batch is failed during the validation, then what will you do for completion of validation?
•
When a quality parameter fails with respect to the specification, a deviation report shall be raised and the investigation shall be conducted immediately for the identification of failure.
•
If the reason for failure is identified, one more consecutive batch shall be considered for the validation run by taking preventive actions to avoid those failures (If necessary revise the MPCR and BPCR).
•
If the reason is unidentified, another three consecutive batches shall be taken for validation
[ads id="ads1"]
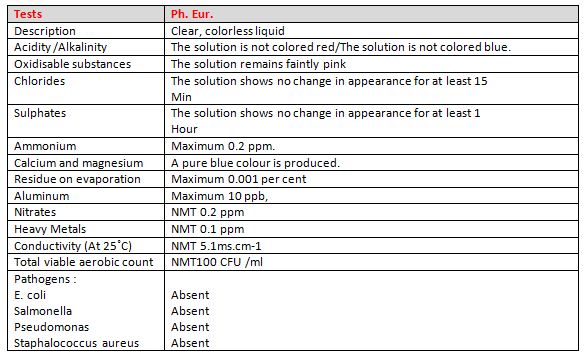
55. What are specifications of Purified water as per any pharmacopoeia?
56. Write the different storage conditions as per any guidelines (specify the name of guideline)?
The different storage conditions are given below as per USP:
•
Freezer: -25°C to -10°C
•
Cold: Any temperature not exceeding 8°C
•
Refrigerator: Between 2°C and 8°C
•
Cool: 8°C to 15°C
•
Room temperature: The temperature at prevailing working area.
•
CRT: 20°C to 25°C
•
Warm: 30°C to 40°C
•
Excessive heat: Above 40°C
[ads id="ads1"]
57. What is ISO 9001, ISO 14001, ISO 18001, and ISO 22001?
•
ISO 9001 : Quality Standard Management
•
ISO 14001 : Environmental Standard Management
•
ISO 18001 : Safety & Health Standard Management
•
ISO 22001 : Hazop Standard Management
[ads id="ads1"]
58. What is HACCP, OHSAS?
•
HACCP: Hazard Analysis Critical Control Point
•
OHSAS: Occupational Health & Safety Assessment Series
59. Why one liter of water is equivalent to one kilogram of water at room temperature?
•
Because of at normal room temperature is between 25°C and 35°C at plant operating condition and the variation in weight Vs. Liter of water is negligible compared to volume.
[ads id="ads1"]
60. What is calibration?
The demonstration that a particular instrument or device produces results within specified limits by comparison with results produced by a reference or traceable standard over an appropriate range of measurements
61. What is the maximum time allowed after cleaning with water as last rinse?
Equipment should not be left with water it after cleaning. The last step of the cleaning procedures involve drying with solvent or flushing with nitrogen, thus ensuring that there is no opportunity for microbial growth.
62. What is the efficiency of the High Efficiency Particulate Air (HEPA) filter?
This type of air filter can remove at least 99.97% particles in air up to 0.3μm in diameter.
63. What is the micron size of HEPA filter?
The micron size of HEPA filter is 0.3μm
64. Do you have any idea about schematic diagram of HVAC system?
65. If two different products are manufacturing in two modules of one production block, then will you accept the common air handling unit for both pharma area? Write “Yes” or “No” with reason?
No, because of cross-contamination (if re-circulation of return air)
Yes, if 100% of fresh air is circulated through the respective area.
[ads id="ads1"]
66. Why blending validation is required? What quality parameters of product are considered for validation and what parameters of equipment are to be considered during validation?
Because of to provide sufficient documented evidence to assure that the blending operation of product is capable of repeatedly and reliably producing a homogeneous material to meet established specifications when operated under defined standard conditions.
The following Quality parameters are to be considered, but not limited:
a) Loss on Drying / Water content
b) Bulk density / tapped density
c) Residual solvent
d) Particle size
The following parameters are to be considered for the equipment during validation, but not limited:
a) Blender capacity
b) RPM of the blender
c) Occupancy of the blender
d) Number of individual batches to be taken for each blend
e) Mixing time
67. What is the formula for calculation of “Air changes per hour” during HVAC validation?
Air changes per hour= [Total CFM of the blower/Filter x 60]/ Total room volume
[ads id="ads1"]
68. During the performance qualification in the vacuum tray dryer, how many temperature probes used?
Total 16 to 24 temperature probes are to be kept during the performance qualification of the vacuum tray dryer (or number of probes specified in the protocol)
69. What is the formula for the calculation of “MACO” while cleaning between one API to another API?
MACO = [Minimum therapeutic dosage of previous product X Minimum batch size of next product] / [Safety factor X Maximum therapeutic dosage of the next product]
70. What is the limit for “Individual unknown Impurity” in API as per ICH Q2A?
The limit of the “Any individual unknown Impurity” is not more than 0.1%
71 What is the Pharmaceutical industry?
The pharmaceutical industry is a sector of the economy that discovers, develops, produces, and markets drugs and other pharmaceutical goods.
The goal of the pharmaceutical industry is to create safe and effective medications to help patients cure or prevent disease.
72. What are the different types of pharmaceutical companies?
Types
- Drug development
- Drug manufacturing
- Research-based pharmaceutical companies
- Generic drug manufacturers
- Contract research organizations (CROs)
- Oncological formulations
- Biotechnology companies
- Specialty pharmaceutical companies.
73. What is the role of a qualified person (QP) in the pharmaceutical industry?
A QPs are legally responsible for certifying batches of medicinal products before they're used in clinical trials or available on the market.
Qualified person (QP) is a technical term used in European Union pharmaceutical regulation.
74. What qualifications are required to become a qualified person?
To become a Qualified Person, one must hold a degree in pharmacy, chemistry, or a related scientific discipline.
You must also have experience working in a pharmaceutical quality environment, including at least two years as a Qualified Person trainee.
75. What is Good Distribution Practice (GDP)?
Good Distribution Practice (GDP) ensures that medicinal products are stored, transported, and distributed in a manner that maintains their quality and integrity.
76. What are the key regulatory bodies governing the pharmaceutical industry?
Key regulatory bodies governing the pharmaceutical industry
Food and Drug Administration (FDA) in the United States (USFDA)
European Medicines Agency (EMA) in Europe
World Health Organization (WHO) globally
77. What is a Drug Master File (DMF)?
A Drug Master File (DMF) is a confidential document submitted to regulatory authorities.
It contains detailed information about the manufacturing, processing, and components of a drug product.
78. What is a Certificate of Analysis (COA)?
A Certificate of Analysis (CoA) is a document provided by a manufacturer that certifies the quality and purity of a batch of pharmaceutical product.
79. What is a pharmacopoeia?
A pharmacopoeia is a book or set of standards that contains information about the quality, purity, and strength of drugs and medications.
80. What is the difference between a brand-name drug and a generic drug?
A brand-name drug is a medication that is marketed under a specific brand name by the company that developed it. A generic drug is a copy of the brand-name drug that contains the same active ingredients and is usually sold at a lower price.
81. What is the Drug Approval Process?
The Drug Approval Process refers to the series of steps that a pharmaceutical product must go through to gain regulatory approval for marketing and sale.
82. What is the role of clinical trials in drug development?
Clinical trials are conducted to evaluate the safety and efficacy of new drugs or treatments in humans before they can be approved for use.
83. What are the different phases of clinical trials?
Clinical trials typically have four phases:
Phase 1 (safety and dosage)
Phase 2 (efficacy and side effects)
Phase 3 (large-scale effectiveness)
Phase 4 (post-marketing surveillance).
84. What is a placebo?
A placebo is an inactive substance or treatment given to participants in a clinical trial to compare the effects of the investigational drug with those of no treatment.
85. What is Good Clinical Practice (GCP)?
Good Clinical Practice (GCP) is an international ethical and scientific quality standard for designing, conducting, recording, and reporting clinical trials.
86. What is a Pharmacovigilance System?
Pharmacovigilance System is responsible for monitoring and collecting information about the A safety and side effects of medicinal products after they have been authorized for use.
87. What is a Risk Management Plan (RMP)?
A Risk Management Plan (RMP) outlines the measures taken to identify, characterize, and minimize the risks associated with a medicinal product throughout its lifecycle.
88. What is a Quality Management System (QMS)?
A Quality Management System (QMS) is a set of policies, processes, and procedures used to ensure that products and services meet quality requirements.
89. What is Process Validation?
Process Validation is the documented evidence that a process consistently produces a result or product meeting predetermined specification
90. What is the role of a Quality Assurance (QA) department in pharmaceutical manufacturing?
The Quality Assurance (QA) department is responsible for ensuring that all manufacturing processes and procedures comply with regulatory requirements and quality standards.
91. What is the role of a Regulatory Affairs (RA) department in the pharmaceutical industry?
The Regulatory Affairs (RA) department is responsible for ensuring compliance with regulatory requirements and managing interactions with regulatory authorities.
92. What is a Batch Record?
A Batch Record is a document that provides a complete history of a batch of pharmaceutical product, including manufacturing, testing, packaging, and distribution information.
93. What is a Deviation?
A Deviation is an unplanned or unanticipated event or occurrence that deviates from established procedures or specifications.
94. What is a Change Control System?
A Change Control System is a process used to manage and document changes to validated systems, processes, or procedures to ensure they are implemented in a controlled manner.
95. What is an Out-of-Specification (OOS) result?
An Out-of-Specification (OOS) result refers to a test result that falls outside the predetermined
acceptance criteria or specifications.
96. What is a Quality Risk Management (QRM) process?
A Quality Risk Management (QRM) process is a systematic approach to identifying, assessing, and controlling risks to product quality, patient safety, and data integrity.
97. What is a Stability Study?
A Stability Study is conducted to evaluate the effect of environmental factors such as temperature, humidity, and light on the quality and stability of a pharmaceutical product over time.
98. What is a Batch Release?
Batch Release refers to the process of reviewing and approving a batch of pharmaceutical product by a qualified person before it can be released for distribution or sale.
99. What is a Recall?
A Recall is the process of removing or correcting marketed pharmaceutical products that are found to be defective, potentially harmful, or in violation of regulatory requirements.
100. What is a Quality Agreement?
A Quality Agreement is a formal document that defines the responsibilities, processes, and
procedures related to quality between two parties involved in the supply chain, such as a
manufacturer and a contract testing laboratory.
101. What is Data Integrity?
Data Integrity refers to the completeness, accuracy, consistency, and reliability of data throughout its lifecycle, including creation, processing, storage, and retrieval.
102. What is a Quality Risk Management (QRM) tool?
A Quality Risk Management (QRM) tool is a systematic approach or software used to assess and manage risks related to product quality, patient safety, and data integrity.
103. What is a Pharmacokinetic Study?
A Pharmacokinetic Study evaluates how drugs are absorbed, distributed, metabolized, and excreted by the body to determine their
pharmacokinetic properties.
104. What is a Pharmacodynamic Study?
A Pharmacodynamic Study evaluates the effects of drugs on the body, including their mechanism of action, efficacy, and safety.
105. What is a Quality Control (QC) laboratory?
A Quality Control (QC) laboratory is responsible for testing and analyzing raw materials, in-process samples, and finished products to ensure they meet quality specifications.
106. What is a Stability Chamber?
A Stability Chamber is an environmental chamber used to store pharmaceutical products under controlled conditions of temperature and humidity to evaluate their stability over time.
107. What is a Process Analytical Technology (PAT)?
Process Analytical Technology (PAT) refers to the use of advanced analytical tools and techniques to monitor and control manufacturing processes in real-time for improved product quality.
108. What is a Risk Assessment?
A Risk Assessment is a systematic process of identifying, evaluating, and prioritizing risks to
determine the best course of action for risk mitigation.
109. What is a Quality Audit?
A Quality Audit is an independent review or examination of processes, procedures, and systems to ensure compliance with regulatory requirements and quality standards.
110. What is a Quality Agreement?
A Quality Agreement is a formal document that defines the responsibilities, processes, and
procedures related to quality between two parties involved in the supply chain, such as a
manufacturer and a contract testing laboratory.
111. What is a Change Control System?
A Change Control System is a process used to manage and document changes to validated systems, processes, or procedures to ensure they are implemented in a controlled manner.
112. What is an Out-of-Specification (OOS) result?
An Out-of-Specification (OOS) result refers to a test result that falls outside the predetermined
acceptance criteria or specifications.
113. What is a Quality Risk Management (QRM) process?
A Quality Risk Management (QRM) process is a systematic approach to identifying, assessing, and controlling risks to product quality, patient safety, and data integrity.
114. What is a Stability Study?
A Stability Study is conducted to evaluate the effect of environmental factors such as temperature, humidity, and light on the quality and stability of a pharmaceutical product over time.
115. What is a Batch Release?
Batch Release refers to the process of reviewing and approving a batch of pharmaceutical product by a qualified person before it can be released for distribution or sale.
116. What is a Recall?
A Recall is the process of removing or correcting marketed pharmaceutical products that are found to be defective, potentially harmful, or in violation of regulatory requirements.
117. What is a Quality Agreement?
A Quality Agreement is a formal document that defines the responsibilities, processes, and
procedures related to quality between two parties involved in the supply chain, such as a
manufacturer and a contract testing laboratory.
118. What is Process Validation?
Process Validation is the documented evidence that a process consistently produces a result or product meeting predetermined specifications
119. What is a Quality Risk Management (QRM) tool?
A Quality Risk Management (QRM) tool is a systematic approach or software used to assess and manage risks related to product quality, patient safety, and data integrity.
120. What is a Pharmacokinetic Study?
A Pharmacokinetic Study evaluates how drugs are absorbed, distributed, metabolized, and excreted by
the body to determine their pharmacokinetic properties.
121. What is a Quality Control (QC) laboratory?
A Quality Control (QC) laboratory is responsible for testing and analyzing raw materials, in-process samples, and finished products to ensure they meet quality specifications.
122. What is a Stability Chamber?
A Stability Chamber is an environmental chamber used to store pharmaceutical products under controlled conditions of temperature and humidity to evaluate their stability over time.
123. What is a Process Analytical Technology (PAT)?
Process Analytical Technology (PAT) refers to the use of advanced analytical tools and techniques to monitor and control manufacturing processes in real-time for improved product quality.
124. What is a Risk Assessment?
A Risk Assessment is a systematic process of identifying, evaluating, and prioritizing risks to
determine the best course of action for risk mitigation.
125. What is a Quality Audit?
A Quality Audit is an independent review or examination of processes, procedures, and systems to ensure compliance with regulatory requirements and quality standards.
126. What is a Quality Management System (QMS)?
A Quality Management System (QMS) is a set of policies, processes, and procedures used to ensure that products and services meet quality requirements.
127. What do you mean by drug product specifications?
A specification is a list of tests, test methods, and limits (like numbers or ranges) that a drug must meet to be considered safe and effective. It tells us what is acceptable for a drug's quality.
If a drug passes all the listed tests using the approved methods, it is said to "conform to specifications."
Specifications are very important. They are part of the drug’s quality control system. These standards are proposed by the drug manufacturer and approved by health authorities before the drug can be sold.
Why Are Specifications Important?
Specifications help ensure:
- The drug is made consistently.
- The drug is safe and works as intended.
- The quality of the drug is maintained over time.
They are just one part of a larger system that includes:
- Testing the raw materials.
- Checking the drug during and after manufacturing.
- Ensuring clean and properly equipped facilities.
- Following Good Manufacturing Practices (GMP).
- Doing stability testing to see how long the drug stays effective.
Release vs. Shelf-Life Specifications
For finished drug products, there may be two sets of limits:
- Release Specifications – Stricter limits used when the drug is first made.
- Shelf-Life Specifications – Slightly broader limits, used throughout the product's life (until its expiry date).
This is done because some small changes (like minor increases in impurities) can happen over time, and these are expected.
- In the US and Japan, official (regulatory) limits are the same at release and at the end of shelf life, but manufacturers may use stricter internal limits at release.
- In the EU, regulators require different limits for release and shelf-life.
128. What Are In-Process Tests?
In-process tests are checks done during the manufacturing of a drug (either the drug substance or the final drug product), not after the product is fully made.
Why Do In-Process Tests Matter?
These tests help make sure that:
The manufacturing process is working correctly.
The product will meet quality standards by the end.
When Are In-Process Tests Not Part of the Official Specification?
Some in-process tests are only used to adjust the process, not to check final product quality. For example:
Measuring tablet hardness or weight before coating.
Checking friability (how easily a tablet breaks).
These kinds of tests are not included in the official specifications because they are just for process control.
When Can In-Process Tests Count Toward the Final Quality Checks?
Some in-process tests can replace final release tests — but only if:
They check the same things (like pH).
They meet the same or stricter limits as the final product test.
It’s been proven (validated) that results won't change between the in-process stage and the finished product.
129. What are the ICH Q guidelines?
1. ICH Q1A (R2) - stability testing of new drug substances and products.
2. ICHQ1B-stability testing: photo stability testing of new drug substances and products.
3. ICH Q1C- stability testing for new dosage forms.
4. ICH Q1D- bracketing and matrixing design for stability testing of new drug substances and
products.
5. ICH Q1E- evaluation of stability data.
6. ICH Q2 (R1)-validation of analytical procedures: text and methodology.
7. ICHQ3A (R2)-impurities in new drug substances.
8. ICHQ3B (R2)-impurities in new drug products.
9. ICHQ3C (R5)-impurities: guidelines for residual solvents.
10. ICHQ3D-guidelines for elemental impurities.
11. ICHQ4B-pharmacopeias
12. ICHQ5A (R1)-viral safety evaluation of biotechnology products derived from cell lines of
human or animal origin.
13. ICHQ5b- analysis of the expression construct in cells used for production of R-DNA
derived protein products.
14. ICHQ5C-stability testing of biotechnological/biological products.
15. ICHQ5D- derivation and characterization of cell subtracts used for production of
biotechnological/biological products.
16. ICHQ5E-comparability of biotechnological/biological products subject to changes in their manufacturing process.
17. ICHQ6A-specifications: test procedure and acceptance criteria for new drug substances and
new drug products: chemical substances.
18. ICHQ6B-specification: test procedures and acceptance criteria for biotechnological /biological products.
19. ICHQ7-good manufacturing practice guide for active pharmaceutical ingredients.
20. ICHQ8 (R2)-pharmaceutical development
21. ICHQ9-quality risk management
22. ICHQ10-pharmaceutical quality system
23. ICHQ11-development and manufacture of drug substances (chemical entities and bio
technological/biological entities.).
24. ICHQ12-technical and regulatory considerations for pharmaceutical product life cycle management.
25. ICHQ13-continous manufacturing of drug substances and drug products.
26. ICHQ14- analytical procedure development
Note: Refer each guidelines for detailed understan
130. What are the stability requirements for drug product submission?
For the general case, the recommended long-term and accelerated storage conditions for Climatic
Zones III and IV are shown below
Study storage condition minimum time period covered by data at submission:
* Long-term 300C ± 20C / 65% RH ± 5% RH 12 Months.
* Accelerated 400C ± 20C / 75% RH ± 5% RH 6 Months.
No intermediate storage condition for stability studies is recommended for Climatic Zone III and
IV. Therefore, the intermediate storage condition is not relevant, when the principle of test
period or shelf life extrapolation described in Q1E are applied.
Aqueous based drug products packaged in semi-permeable containers:
Aqueous based drug products packaged in semi-permeable containers, the recommended long
term and accelerated storage conditions for Climatic Zone III and IV are given below
* Long-term 300C ± 20C / 35% RH ± 5% RH 12 Months.
* Accelerated 400C ± 20C / not more than 25% RH ± 5% RH 6 Months.
131. What do you mean by freeze thaw stability studies?
Freeze-thaw cycle testing is a part of stability testing that allows you to determine if your
formula will remain stable under various conditions.
This type of test puts your sample through a series of extreme, rapid temperature changes that it may encounter during normal shipping and handling processes.
Freeze-thaw stability testing is highly recommended, especially for liquid-
based cosmetics. These products may experience phase separation that can negatively affect the
intended function.
Freeze-thaw testing is conducted by exposing the product to freezing temperatures
(approximately -10 °C) for 24 hours, and then allowing it to thaw at room temperature for 24
hours.
The sample is then placed in a higher temperature (approximately 45°C) for 24 hours and then placed at room temperature again for 24 hours.
The sample is analyzed for significant
changes. This completes one cycle. If, after three cycles of freeze-thaw testing, no significant
changes are observed, you can be confident that the stability of your product is sufficient for
transport.
It is generally conducted in liquid/semi-solid dosage forms.
132. What do you mean by documentation/GDP?
What Is Documentation in Pharma?
In pharmaceuticals, a document is any written record or proof of an activity.
It shows what was done, how it was done, and who did it.
Why Is Documentation Important?
Documentation helps to:
Clearly define how work should be done.
Avoid mistakes caused by unclear or spoken instructions.
Confirm that everything was done correctly.
Allow review, checks, and tracking of a product’s history.
Provide proof to regulatory authorities that the product is made as per standards.
Documents are as important as the product itself.
Regulatory authorities (like WHO, FDA) usually check documents first — before approving the product.
What Is GDP (Good Documentation Practice)?
In pharma, "If it’s not written down, it didn’t happen."
Good documentation protects the patient, the company, and the product.
GDP stands for Good Documentation Practice, which means:
> "Writing, managing, and using documents in a clear, correct, and controlled way to ensure product quality and regulatory compliance."
This includes:
- Preparing
- Checking
- Approving
- Issuing
- Storing
- Reviewing
Example: Batch Manufacturing Record (BMR)
BMR is a key document that shows how each batch of medicine was made.
The product cannot be released unless the BMR is properly reviewed and approved.
BMR is kept along with a sample of the product for 1 year after expiry.
All Pharma Activities Must Be Documented
Every activity (like production, testing, cleaning, etc.) must be written down clearly — usually as SOPs (Standard Operating Procedures).
If something is not documented, it is considered not done.
Controlling Documents
To prevent errors or misuse:
A master copy of each document is made and stamped as "Master Copy" in red.
Photocopies given to departments are marked as "Control Copy."
A logbook is maintained for tracking issued documents (with date and signatures).
Each document should have:
- Effective date
- Review date
- Revision number
Purpose of Documentation
Documentation helps to:
- Define clear steps and specifications for materials and processes.
- Guide staff on what to do and when.
- Provide necessary information for product release.
- Create a traceable audit trail for investigations.
- Provide data for validation, reviews, and analysis.
Types of Pharma Documents
According to Purpose:
1. Specifications – Requirements for quality (for raw materials, packaging, finished products).
2. SOPs – Step-by-step instructions for routine tasks.
3. Records & Reports – Proof of work done (e.g., test results, cleaning records).
4. Logbooks – Continuous records of activities (e.g., equipment use, temperature logs).
5. Protocols – Plans for studies like validation or stability testing.
6. BMR/BPR – Batch Manufacturing/Packaging Records.
Based on Area:
- Personnel & Training
- Buildings & Facilities
- Equipment
- Raw Materials & Packaging
- Production & Process Control
- Packaging & Labeling
- Storage & Distribution
- Laboratory Control
- Returns & Salvaged Products
Factors Affecting Document Design
Documents may vary based on:
1. Type of product (tablet, injection, etc.)
2. Country’s regulatory requirements
3. Use of systems like ERP or SAP
133. What is Batch Manufacturing Record (BMR)?
Batch Manufacturing records: it is an important document issued for every batch of product to
assure, review and record keeping of any product batch.
There are following major content of BMR.
1. Name of product, generic name, strength, shelf life, manufacturing date and expiry date.
2. A complete list of ingredients with full description, codes and quantity to be issued.
3. A statement for theoretical yield and reconciliation.
4. A complete MFG and control instructions, sampling and testing procedure, specification
and precaution to be followed.
5. A statement for processing location and equipment.
6. The method or reference to method to be used for preparing the critical equipment
including cleaning, assembling, calibrating and sterilizing.
7. Dates and time of all activities
8. Line clearance procedure in every steps
9. Labeling control and specimen for coding in primary, secondary and tertiary packing
materials
10. Deviation record
11. Result of examine made.
134. What do you mean by variations /Trend /OOS /OOT?
Variation: means you won’t usually have a perfect linear trend (analytical variability,
sample uniformity)
Trends: if the majority of stations show a trend (downward, upward), consider it a trend.
Out of Trend (OOT): analytical value outside our experience but within the specification
(no OOS)
OOS (Out of Specification): analytical value outside of the registered specification
135. What do you mean by “Significant Change” during stability testing?
For an API: "significant change" is failure to meet the specification for any parameter
For a Finished Pharmaceutical Product (FPP), significant change is any of:
- Any degradation product exceeding its limit
- Failure in tests of appearance, physical attributes and functionality test, e.g. colour, hardness, pH
- > 5% change in assay from initial, i.e. t0 (Initial time point)
- failure to pass dissolution testing for 12 dosage units (fail S2)
136. What do you mean by QbD?
The pharmaceutical Quality by Design (QbD) is a systematic approach to development that
begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.
Quality by Design (QbD) is emerging to enhance the assurance of safe, effective drug supply to the consumer, and also offers promise to significantly improve manufacturing quality performance.
QbD development process includes:
Begin with a target product profile that describes the use, safety and efficacy of the product
Define a target product quality profile that will be used by formulators and process engineers as a quantitative surrogate for aspects of clinical safety and efficacy during product development
Gather relevant prior knowledge about the drug substance, potential excipients and process operations into a knowledge space. Use risk assessment to prioritize knowledge gaps for further investigation
Design a formulation and identify the critical material (quality) attributes of the final
product that must be controlled to meet the target product quality profile.
Design a manufacturing process to produce a final product having these critical materials
attributes.
Identify the critical process parameters and input (raw) material attributes that must be
controlled to achieve these critical material attributes of the final product.
Use risk assessment to prioritize process parameters and material attributes for experimental verification.
Combine prior knowledge with experiments to establish a design space or
other representation of process understanding.
Establish a control strategy for the entire process that may include input material controls, process controls and monitors, design spaces around individual or multiple unit operations, and/or final product tests.
The control strategy should encompass expected
changes in scale and can be guided by a risk assessment.
Continually monitor and update the process to assure consistent quality.
Design of experiments (DOE), risk assessment, and process analytical technology (PAT) are
tools that may be used in the QbD process when appropriate. They are not check-box requirements.
Traditional approach & Enhanced QbD approach
Advantages of QbD
Benefits for Industry:
- Better understanding of the process.
- Less batch failure.
- More efficient and effective control of change.
- Return on investment / cost savings.
Additional opportunities:
An enhance QbD approach to pharmaceutical development provides opportunities for
more flexible regulatory approaches.
Ex: Manufacturing changes within the approved design space without further regulatory
review.
Reduction of post-approval submissions.
Better innovation due to the ability to improve processes without resubmission to the
FDA when remaining in the Design Space.
More efficient technology transfer to manufacturing.
Greater regulator confidence of robust products.
Risk-based approach and identification.
Innovative process validation approaches.
Less intense regulatory oversight and less post-approval submissions.
For the consumer, greater drug consistency.
More drug availability and less recall.
Improved yields, lower cost, less investigations, reduced testing, etc.
Time to market reductions: from 12 to 6 years realized by amongst others.
First time right: lean assets management.
Continuous improvement over the total product life cycle (i.e. controlled, patient guided
variability).
Absence of design freeze (no variation issues).
Less validation burden.
Real time controls (less batch controls).
Realistic risk perceptions.
Contributes substantially to realize the better, cheaper and safer mandate.
137. What do you understand by QTPP (Quality Target Product Profile?
QTPP tells scientists what the medicine should be like (in terms of quality) to make sure it is safe and works well.
The Quality Target Product Profile (QTPP) is like a blueprint or roadmap for developing a drug. It describes the important quality features a medicine should have so that it is safe and effective for patients.
It is created at the beginning of drug development with the final product in mind.
It sets clear goals for the development team to work toward.
It can be updated as new information comes up during research and testing.
QTPP is part of the bigger Target Product Profile (TPP), which focuses on how the medicine should look on the label for patients. QTPP, however, is more focused on the technical side—the chemistry, manufacturing, and quality control of the drug.
138. What do you understand by CQAs (Critical Quality Attrubutes)?
CQAs are the critical features of a drug that must be controlled to make sure the product is safe, effective, and high quality.
Critical Quality Attributes (CQAs) are the key properties of a drug (physical, chemical, biological, or microbiological) that must stay within certain limits so the medicine is safe, effective, and of good quality.
They can be linked to raw materials (like drug substance or excipients), in-process materials, or the final drug product.
CQAs come from the QTPP and help guide product and process development.
They affect important factors like purity, strength, stability, drug release, and safety.
Examples:
Particle size of the drug can affect how the medicine dissolves and releases.
Tablet hardness can influence drug release in controlled-release forms.
139. What do you understand by BE studies?
BE studies check whether a generic drug behaves the same as the brand drug in the body, ensuring patients can safely use it as an alternative.
Bioequivalence (BE) studies are tests done to check if two drug products (usually a brand and a generic) work in the same way inside the body.
If two medicines are bioequivalent, it means they have the same effect on the body in terms of safety and effectiveness.
To prove this, scientists compare how the drug is absorbed into the blood (its bioavailability) using parameters like:
Cmax → the highest concentration of the drug in the blood.
Tmax → the time it takes to reach that highest concentration.
AUC → the total amount of drug absorbed over time.
BE studies are usually done in healthy volunteers under both fasting and fed (after food) conditions.
Regulatory agencies (FDA, WHO, EMA, TGA, etc.) generally accept that if the test (generic) drug’s values fall between 80% and 125% of the reference (brand) drug, the two are considered bioequivalent.
140. How to select blend uniformity samples and what are the acceptance criteria?
1. Carefully identify at least 10 sampling locations in the blender to represent potential areas of poor blending. For example, in tumbling blenders (such as V-blenders, double cones, or drum mixers), samples should be selected from at least two depths along the axis of the blender.
For convective blenders (such as a ribbon blender), a special effort should be made to implement uniform volumetric sampling to include the corners and discharge area (at least 20 locations are recommended to adequately validate convective blenders).
2. Collect at least 3 replicate samples from each location.
Samples should meet the following criteria:
• Assay one sample per location (number of samples (n) ≥ 10) (n = 20 for ribbon blender).
• RSD (relative standard deviation) of all individual results ≤ 5.0 percent.
• All individual results are within 10.0 percent (absolute) of the mean of the results.
141. What do you understand by Uniformity of Dosage Units?
To ensure the consistency of dosage units, each unit in a Suspension, batch should have a drug
substance content within a narrow range around the label claim.
Dosage units are defined as dosage forms containing a single dose or a part of a dose of drug substance in each unit.
The uniformity of dosage units specification is not intended to apply to suspensions, emulsions, or gels in unit-dose containers intended for external, cutaneous administration.
The term “uniformity of dosage unit” is defined as the degree of uniformity in the amount of the drug substance among dosage units.
The uniformity of dosage units can be demonstrated by either of two methods, Content Uniformity or Weight Variation.
Table. Application of Content Uniformity (CU) and Weight Variation (WV) Tests for Dosage Forms
142. How many types of synthesis reactions are used in pharmaceuticals?
In pharmaceutical chemistry, several synthesis reactions are commonly used to build drug molecules. Major types include:
Substitution reactions – replacing one group with another.
Addition reactions – adding atoms/groups to double or triple bonds.
Elimination reactions – removing atoms/groups to form double bonds.
Oxidation–reduction reactions – altering oxidation state of compounds.
Condensation reactions – combining molecules with loss of small molecules (e.g., water).
Hydrolysis reactions – breaking bonds using water.
Rearrangement reactions – shifting atoms within molecules to form new structures.
These are the fundamental reactions used in drug synthesis.
143. What is sterile production?
Sterile production is the manufacturing of products that must be completely free from viable microorganisms and pyrogens. It is critical in pharmaceuticals for products administered directly into the body (e.g., injections, eye drops, IV fluids). Key points are:
Definition: Controlled production ensuring zero microbial contamination.
Environment: Performed in aseptic/cleanroom areas with strict controls.
Techniques: Includes sterilization methods (filtration, autoclaving, dry heat) and aseptic processing.
Examples: Parenteral drugs, vaccines, ophthalmic solutions, implants.
Importance: Ensures patient safety, product quality, and regulatory compliance.
Sterile production = contamination-free manufacturing of sensitive pharmaceutical products.
Q. How do you check equipment cleaning record?
Keep it systematic and confident.
Show understanding of GMP and documentation practices.
Emphasize accuracy, traceability, and compliance.
1. Verify record completeness:
Ensure all required fields are filled — equipment name, ID, date, time, and operator’s signature.
2. Check cleaning status:
Confirm whether it’s marked as ‘Cleaned’, ‘To be Cleaned’, or ‘In Use’ as per status label or logbook.
3. Cross-check with SOP:
Verify cleaning steps and frequency are followed as per the approved SOP or Cleaning Validation Protocol.
4. Review cleaning agent and batch details:
Ensure correct cleaning materials, concentrations, and lot numbers are recorded.
5. Verify inspection and approval:
Confirm that the cleaning has been checked and signed by QA or responsible supervisor before next use.
6. Check for traceability:
Ensure record links to previous and next product batches to avoid cross-contamination.
Example Closing Line:
“I ensure every cleaning record is complete, verified as per SOP, and approved before equipment release — maintaining compliance and product safety.”
Thanks for reading -Pharmaceutical Interview Questions
Naitik Patel
Industrial Guide